
Publicado em 8 de Julho de 2014 às 10:43 em: Bioinformática
Dando continuidade a nossa série de artigos sobre sequenciamento clínico, neste post vamos nos aprofundar tecnicamente sobre os formatos utilizados para representar as sequências de DNA de um paciente, e saber como esses dados são armazenados. A partir destes dados é que nós da Genomika conseguimos minerar, buscar, identificar mutações (variantes) que possam estar associadas a um fenótipo clínico de uma doença em investigação.
O primeiro passo é compreender como digitalizamos seu DNA, analogamente a um scanner que escaneia documentos físicos em papel para um formato digital legível como um PDF, Word, etc. O processo de digitalização de DNA, que tecnicamente denominamos de sequenciamento de DNA, é realizado em várias etapas, desde processos bioquímicos a processamento de software para armazenar o DNA físico real em formato eletrônico formado por uma sequência de bases A, T, C, G. Os sequenciadores de segunda geração conseguem gerar milhões de fragmentos (reads) em maior velocidade e menor tempo, em comparação aos fragmentos produzidos pela tecnologia Sanger (sequenciadores mais antigos, mas ainda muito utilizados). Aqui entram os algoritmos de bioinformática, que alinham estes fragmentos em relação a um arquivo de referência, o nosso genoma por exemplo. Essa digitalização facilita as tarefas de busca, armazenamento, transporte e análise destes dados.
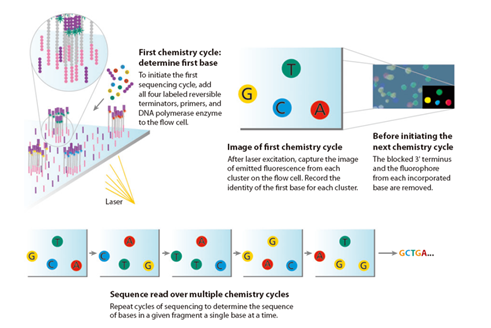
E como guardamos estas sequências de bases ? Dois formatos se tornaram padrões universais atualmente: FASTA e FASTQ. Um arquivo FASTA contém um ou mais sequências de nucleotídeos de DNA. É um formato simples, que faz as sequências serem fáceis de analisar. Cada sequência tem 1 linha de cabeçalho com informações sobre a sequência (por ex: um identificador), seguido por uma ou mais linhas de bases lidas do DNA. O FASTA também pode ser usado para representar os aminoácidos que formam uma proteína, e sequências de RNA.
>Taxon1 CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC >Taxon2 CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT >Taxon3 CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC
Adicionalmente, os sequenciadores da Genomika geram um arquivo FASTQ. FASTQ ? Sim, este também é outro formato universal que vem ganhando muita popularidade na comunidade científica e fabricantes de sequenciadores . É um arquivo similar ao FASTA, porém inclui informações importantes sobre a qualidade de sequenciamento daquela sequência. Ilustramos um exemplo a seguir:
@HWI-ST999:102:D1N6AACXX:1:1101:1235:1936 1:N:0: (IDENTIFICADOR) ATGTCTCCTGGACCCCTCTGTGCCCAAGCTCCTCATGCATCCTCCTCAGCAACTTGTCCTGTAGCTGAGGCTCA CTGACTACCAGCTGCAG (SEQUENCIA) + 1:DAADDDF\<B\<AGF=FGIEHCCD9DG=1E9?D>CF@HHG??B\<GEBGHCG;;CDB8==C@@>>GII@@5? A?@B>CEDCFCC:;?CCCAC (QUALITY SCORE)
Esses escores de qualidade são extremamente importantes para analisarmos variantes e mutações. Sequências de baixa qualidade podem ser automaticamente eliminadas da análise, aumentando a confiabilidade nos resultados. Na Genomika nos preocupamos com o controle de qualidade para que possamos entregar ao paciente e médico solicitante os resultados mais fidedignos a partir do seu DNA, e usamos estas informações para uma pré-análise e triagem.
Agora que você compreendeu como armazenamos seu DNA eletronicamente, ficará mais fácil de entender os próximos passos de nossos exames de sequenciamento clínico. Como vocês perceberam, a bioinformática desempenha um papel importante em nossos exames.
Algumas curiosidades:
1) Há um banco mundial de sequências de vários organismos, se você por exemplo abrir este FASTA, vai concluir que ele é o genoma da bactéria Xylella fastidiosa, o primeiro organismo com genoma totalmente decifrado por uma rede de cientistas brasileiros e que causa a doença chamada amarelinho em laranjas.
2) O fasta do genoma humano, por exemplo na versão HG19; possui 3GB com mais de 62.743.362 linhas.